
Conditions for enhancement of gas phase chemical reactions inside a dark microwave cavity

Let us focus here on a collision of atom A with diatom BC (reactants) to form a finite lifetime activated complex [ABC]#, and then proceeds to yield the products AB + C. Reaction rates16 and its cumulative reaction probabilities17 can be calculated by the standard (Hermitian) quantum scattering theory. In particular, ref. 17 presents the transition from Lippmann-Schwinger formula for transition probability to Flux operator, calculated at any given divided surface. In standard quantum scattering theory, the Green operator is defined as \(\hatG^+(E)=\lim _\epsilon \to 0(E+i\epsilon -\hatH_ABC)^-1\) where \(\hatH_ABC\) is the real physical Hamiltonian for the A + BC → AB + C reaction. Notice that the term iϵ in the Green operator introduces outgoing wave boundary condition into \(\vert \Phi \rangle =\hatG^+(E)\vert \phi \rangle\) (see solution to exercise 10.1 in ref. 15). Thus, the Green operator is a non-Hermitian operator. Let us quote from ref. 18: “The branching of quantum mechanics to standard (Hermitian) formalism and non-Hermitian (NH) formalism is associated with the decision to express the exact energy spectrum with one of the two possible self-consistent like problems where the use of the Green operator imposes an outgoing boundary condition on the solutions of the time-independent Schrödinger equation.” Within the framework of the NHQM formalism the Green operator is described using all the discrete complex poles of the scattering matrix. That is, the NHQM Green operator, in its spectral representation, is defined as \(\hatG(E)=\sum _pol\frac\left\vert E_pol\right)\left(E_pol \right\vert E-E_pol\), were Epol is a complex number. Note that here we expand the non-Hermitian Green operator in a basis set of the complex poles of the Hamiltonian, and we should thus use the complex inner product (so-called c-product15) rather than the scalar product that is commonly used within the Hermitian formalism. Namely, \(| E_pol\left)\right.=\vert E_pol^* \rangle\).
Not all complex poles in NHQM are linked to resonance states; nevertheless, all resonances are indeed complex poles of the scattering matrix, which we designate as physical complex poles. These physical and non-physical discrete complex poles can serve as a basis set for calculating reaction rates for structure-less transition probabilities across a potential barrier19. However, in this paper, we only use the physical ones, which are divided into two categories. The occupied resonance, a transition state (TS), which corresponds to a predissociation of the activated complex, as described above. The TS resonance is associated with the reaction that takes place outside of the cavity (when the two mirrors are sufficiently far apart) and can be described by a classical mechanism where the reactants have enough energy to overcome the potential barrier of the reaction. The other physical resonances decay faster than the TS resonance due to the trapping of reactants for a relatively extended time period due to bound vibrations perpendicular to the reaction coordinate near the saddle point in the potential energy surface. Thus, even in the context of gas phase reactions, the relatively long lifetime of the activated complex arises due to crucial role played by Feshbach resonances, which result from the dynamics in the two-dimensional space. To the just discussed effective potential energy barrier that results from the dynamics in 2D space we will refer later as a dynamical potential barrier (DB). As we will explain below, the calculations of these physical resonances can be simplified by using the nuclear adiabatic approach. This is done in order to avoid the calculations of the resonance solutions of a multi-dimensional nuclear time-independent Schrödinger equation by calculating the resonances of a modified adiabatic one-dimensional (1D) problem. Since these physical resonances are often embedded inside the 1D potential barrier (of the effective 1D adiabatic potential) we refer to them as dynamical barrier (DB) resonances. For further explanation see ref. 20.
The TS predissociation resonance of the activated complex [ABC]# can be coupled to one of the DB resonances with a quantized field mode. The TS resonance is the only one associated with the vacuum of the cavity and therefore is the only physical resonance that is occupied before tuning the distance between the two mirrors of the cavity to couple the occupied TS state with one of the selected DB resonances. Before delving into the cavity effect on chemical reactions, we must first describe the nuclear adiabatic theorem obtained from the solution of the time-independent Schrödinger equation using the reaction path Hamiltonian.
In the nuclear adiabatic theorem, it is assumed that the motion of the products (as they collide to create the activated complex and yield the chemical reaction products) is much slower along the reaction coordinate compared to the motion in the perpendicular directions to the reaction coordinate21,22. As we will show below the nuclear adiabatic theory enables us to simplify the calculations of the predissociation resonance and dynamical barrier states.
The reaction path Hamiltonian from a potential energy surface
The potential in multi-dimensional space in mass weighted coordinates is given by V(X, Y1, Y2, …, Y3N−6), where X is the reaction coordinate and \(\Y_j\_j = 1,2\ldots ,3N-6\) are perpendicular coordinates to X. Therefore, (X = 0, Y = 0) is a saddle point, which is associated with a transition state as determined by electronic structure calculations. Here, N is the number of atoms in the A + B (incoming channel), which equals to the number of atoms in C + D (outgoing channel), and 3N − 6 is the number of internal degrees of freedom. However, only 3N − 7 degrees are relevant since one degree corresponds to the reaction direction. The frequency of a transition state along the reaction has an imaginary value and for the reactant/products it is zero since it corresponds to the vibration between (minimum) two species that are far apart. Therefore, approximately, we can describe this reaction path Hamiltonian (RPH) in the vicinity of the reaction coordinate where,
$$V(X,Y_1,Y_2,\ldots ,Y_3N-6) \, \approx V_r.c(X)+\sum_j=1^3N-7\frac\mu \omega _j^2(X)2Y_j^2,$$
(1)
where μ = mAmBC/(mA + mBC) is the reduces mass and Vr.c.(X), the 1D potential (corresponding to the electronic many-body solutions at different X) calculated along the reaction coordinate supports a potential barrier at X ≡ 0. The predissociation resonance solutions are the eigenfunctions of \(\hatH_RPH\) when outgoing wave boundary conditions are imposed. The narrowest predissociation resonance with energy which is approximately as the energy in classical trajectory calculations that describe the reaction from reactants to product is associated with the transition state. Here we get into an important issue in our study. Since the TS is associated with the narrowest predissociation resonance all other resonances decay faster. The ability to couple the TS with one of the broader resonance (denoted here as DB resonance) in a dark cavity depends on the ability to have a quantized field mode ℏωcav = Re[ETS] − Re[EDB] > 0. This condition is crucial for enhancement of rates of reaction in a dark cavity.
Using the adiabatic approximation of the spectrum (bound and continuum states of complex poles when the outgoing boundary conditions are applied) this RPH is reduced into 1D effective Hamiltonian
$$\hatH_ad=\frac\hatp_x^22+V_ad(X)$$
where
$$V_ad(X)=V_r.c(X)+\hslash \sum_j=1^3N-7\omega _j(X)\left(n_j+\frac12\right)\equiv V_SB(X)+V_DB(X)$$
(2)
For nj = 0 we get the adiabatic Hamiltonian for the ground vibrational states of the normal modes perpendicular to the reaction coordinate. In the adiabatic potential given in Eq. (2) there are two type potential barriers. A static potential barrier (SB), VSB(X), is associated with Vr.c.(X). The dynamical potential barrier (DB), VDB(X), results from the bound vibrational modes that are perpendicular to the reaction coordinate X. The sum of these provides the effective 1D adiabatic potential. The complex poles that will be obtained in the next section are the Feshbach resonances that would be obtained from the study of the dynamics of the multi-dimensional PES as given in Eq. (2). This approach has been previously used to study the collisions of hydrogen atom with ArCl Van der Waals molecule20 and for several collision reactions, such as H + H223,24. Notice that the electronic potential surface does not depend on the mass of the nuclei, and therefore it is the same for any isotope that is used in the experiments. The use of deuterium rather of hydrogen is desirable in order to increase the number of Feshbach type predissociation resonances. It’s important to note that the complex physical poles are embedded inside the dynamical potential barrier that is obtained due the use of the adiabatic approximation described above, and they are not embedded in the so-called static potential barrier of Vr.c.(X). This is a crucial point in our derivation of the conditions where the quantized field modes in a dark cavity are not excited (i.e., in its vacuum field state).
Following our theoretical approach to enhance the reaction rate within the cavity, we must ensure that the dynamical potential barrier supports complex poles which are not obtained (even not approximately) from the static potential barrier. Let us discuss the following reactions H + ArCl → [ArHCl]# → H + Ar + Cl and O + D2 → [ODD]# → OD + D as examples for reactions that can be enhanced by a dark cavity.
The enhancement of the rate of the reaction H + ArCl → [ArHCl]# → H + Ar + Cl
This reaction has been studied in detail in ref. 20 in free space (when the distance between the two mirrors is taken to be infinitely large). In this work, it has been shown that the predissociation resonances of [ArHCl]# which results from the vibrations perpendicular to the reaction coordinate (when hydrogen atom oscillates between Ar and Cl) are accurately obtained from the 1D effective potential barrier that is obtained within the framework of the nuclear adiabatic approximation. Here we wish to show that in this case where all complex poles are associated with the dynamical potential barrier and are physical poles the rate of the reaction is enhanced by the dark cavity that couples the TS resonance with one of the DB resonance of ArHCl. The physical poles are associated with the Feshbach resonances of the 2D potential energy surface, and their associated energies (the real part of the complex energy of the pole of the scattering matrix) are larger than the threshold energy of the 1D effective potential. All other poles are non-physical and can only be used as part of the discrete basis set constructed from all complex poles of the scattering matrix. A simple distinction between the physical and non-physical complex poles can be made by comparing the complex poles associated with shape-type resonances obtained from the 1D potential as a function of the reaction coordinate against the complex poles of the adiabatic effective 1D potential energy curve. The complex poles of the adiabatic 1D potential energy curve approximate the 2D Feshbach resonances mentioned above. The poles associated with the 1D shape-type resonances are false resonances and they are very different from the Feshbach resonances obtained from the 2D reaction path Hamiltonian, which are considered the physical complex poles.
In Fig. 1 we show the dynamical potential barrier for the reaction
$$H+ArCl\to [ArHCl]^\#\to H+Ar+Cl$$
and the positions of the TS and DB resonances. The potential barrier is symmetric although the reaction is asymmetric (products not equal to reactants) due to the weak van der Waals bonds of ArCl whereas the bonds in the activated complex [ArHCl]# are stronger, which results in negligible static potential barrier along the reaction coordinate in comparison to the dynamical potential barriers. In Fig. 2 the predissociation resonances of ArHCl are presented counted by the number of their nodes. The TS and the DB resonance that would be coupled by the dark cavity are marked by a blue color. The TS resonance state is localized at the top of the potential barrier, it has the longest lifetime and it looks similar to the Gaussian wavefunction as obtained for a parabolic potential barrier25.

The potential is adopted from ref. 20 and the resonance positions are calculated herein using complex scaling. Potential in Hartree and coordinate also in atomic units. These resonances are actually the dynamical resonances as obtained from the full multi-dimensional calculations, notice that the static potential barrier along the reaction coordinate is negligible (see ref. 20).

All of them results from the vibrational states of ArHCl when along the reaction coordinate H has almost a free translational motion with respect to the ArCl center of mass. The number of nodes were counted from the plots of the square integrable complex scaled ∣Ψres(X)∣2. Notice that the transition state is associated the longest lifetime resonance that is localized at the top of the potential barrier. The cavity is tuned to couple the two states marked in blue. Energies in Hartree.
Below in Fig. 3, we show the enhancement of the rate of this reaction when the distance between the two mirrors in the Fabry–Perot dark cavity are tuned to have a quantized field mode with the frequency ωcav = 0.195 meV. This quantized field mode corresponds to the resonance condition that couples the TS resonance and the DB resonance, which are marked in blue in Fig. 2.
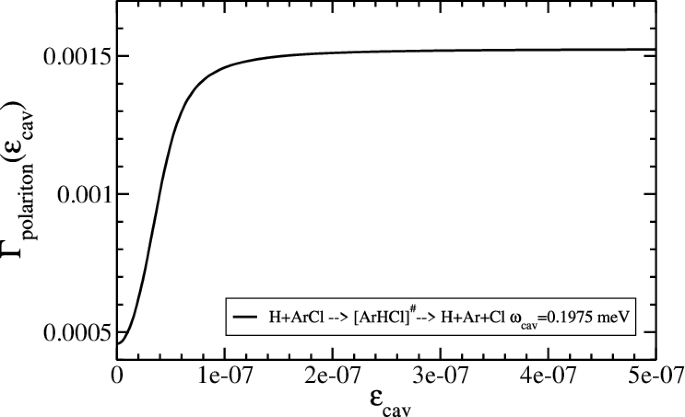
The cavity can be tuned into this resonance condition by adjusting the distance between the two Fabry–Perot mirrors (see Eq. (6)). Detailed of the calculation are given in the section “Results and discussion”, in particular, Eq. (13). Rate in Hartree and coupling in atomic units. Notice that \(E_TS-E_DB(3nodes)\approx \hslash \omega _cav(1_photon+\frac12)\). Here we used such a distance between the two mirrors that generates one mode resonant with the molecular transition (i.e., nphoton = 1) as mentioned in ref. 13. The enhancement is saturated quickly, since in this case the probabilities of the upper and lower polaritons to populate the transition state become about equal as ϵcav is increased. Roughly speaking, the rate of reaction is about equal to the averaged value of the TS decay rate outside of the cavity and the decay rate of the broad three mode resonance (shown in Fig. 2). This point will be explained later by Eq. (12).
This result encourages experimentalists to study such phenomena in the context of either cold or room temperature molecular collisions in gas phase, as for example in Narevicius Lab26. Generally speaking, the relevant temperatures should be high enough as to allow overcoming classically the static potential barrier. In the particular case discussed here (the collision of deuterium diatomic molecules with oxygen) the experiment has to be done at room temperature. On the other hand, an experiment involving ArHCl should be done preferably at cold temperatures, because the static potential barrier along the reaction coordinate is negligible. In the experiments of ref. 26, controlling the temperature implies to increase the angle between the angle of the two colliding atomic/molecular beams (this is equivalent to increase the temperature of the collision).
The enhancement of the rate of the reaction O + D2 → [ODD]# → OD + D
This reaction represents the most common situation where the chemical bonds in the reactants and the products are stronger than the chemical bonds of the activated complex. In the type of reactions the static potential barrier is much more pronounced than the dynamical potential barriers. The physical predissociation resonances of the activated complex are associated withe complex poles that are obtained from the 1D effective potential obtained within the framework of the adiabatic approximation where the vibrations perpendicular to the reaction coordinate are taken into consideration.
In ref. 14, Johnson and Winter studied the the effect of vibrational energy on the reaction of molecular hydrogen with atomic oxygen. They calculated the the contour map for the \(O+H_2\to ^\Gamma (\epsilon _cav=0)OH+H\), in which O collides with H2. See below their contour map plot as presented in Fig. 1 in their paper. Notice that in the electronic structure calculations the isotopic effects are not taken into consideration and therefore we can replace hydrogen molecule by deuterium diatomic molecule. The product is OD + D and the frequency of the vibration which is perpendicular to the reaction coordinate is denoted by Ωvib(X). The incoming oxygen atoms collide with deuterium diatomic molecules when they are temporarily trapped for a long time around the saddle point due to the oscillations perpendicular to the reaction coordinate. This temporarily trapping results in the formation of a long lived transition state (TS) predissociation resonance. Nevertheless, there are other excited predissociation resonances which are not populated but, as we will show here, will interact with the TS resonance in a dark cavity resulting in enhancement of the reaction. The different of the masses of the isotopes will be taken into consideration in the next step where we will describe potential energy surface for this reaction using the reaction path Hamiltonian representation.
The 3D and 2D potential energy surfaces as described in Fig. 4 were obtained respectively by using Eq. (1) and Eq. (2). The information for this plot comes from Johnson and Winter in 1977, see ref. 14. On this scale, one cannot see the energy barrier in the X-direction. Our focus is on the weaker, “soft” vibrations along the Y-axis, compared to the vibrations in the original reactants (O + D2) and the products (OD + D). In Fig. 5, one can see the static potential barrier along the reaction coordinate. As explained in the section “An intuitive (Hermitian) explanation of the cavity-induced reaction rate enhancement”, the reactants O and D2 approaching one another as they move along X < 0. Due to the relative high frequency of the ground vibrational level of D2 the collision energy is about equal to the height of the static potential barrier. However, as they get to be close enough to form the activated complex [ODD]# the energy transfer from the excited vibrational soft mode of [ODD]# to the X-component of the translational kinetic energy will enable to bypass the potential barrier. Notice that VSB(X) is approximately described as an asymmetric Eckart potential barrier given by (Hartree unites) as a function pf the reaction coordinate given in Bohr unites,
$$V_SB(X)= -0.0180244\,\cosh ^2(0.05)\,[\tanh (X+0.05)-\tanh (0.05)]^2 \\ +0.0180244\times e^2\cdot 0.05$$
(3)

The reaction follows a path (so called reaction coordinate) that goes over a high-energy point called the saddle point, forming a temporary structure of an activated complex labeled [ODD]#. The Y-axis is perpendicular to the direction of the reaction rate coordinate, X. Both axes are measured in Bohr.

As in the 3D plot of the potential surface the focus here is on the weaker, “soft” vibrations along the Y-axis, compared to the vibrations in the original reactants (O + D2) and the products (OD + D). The zoom enables one to see the static potential barrier unlike in Fig. 4.
The static potential barrier VSB(X) is shown in Fig. 6

Potential in Hartree and coordinate in Bohr. Notice that the energy of the reactants, O + D2, is taken as zero.
The VDB(X) = Ωvib(X)(nY = 0 + 1/2) is given by VDB(X) = Ωvib(X)(nvib = 0 + 1/2) where Ωvib(X) is described in Fig. 7.
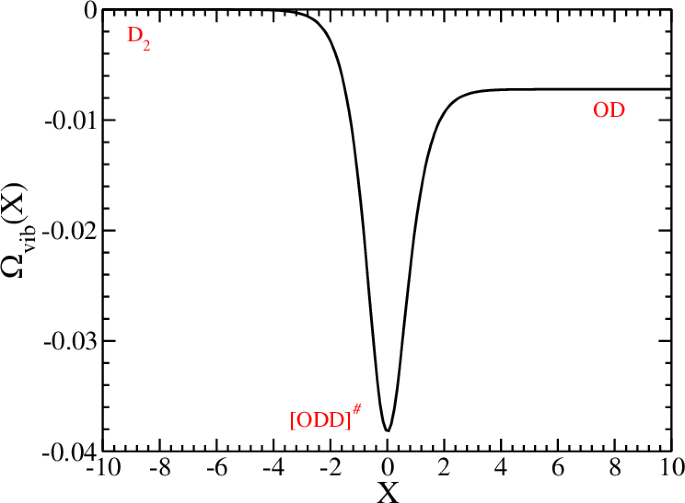
The frequency of the vibration perpendicular (Y) to the reaction coordinate as a function of the reaction coordinate X. Notice that the vibrational frequency of the reactant D2 is taken as a reference and therefore the frequency of the product OD gets a negative value. Frequencies in Hartree and coordinate in Bohr.
The sum VSB(X) + VDB(X) provides the adiabatic potential that includes the effect of vibrational energy on the reaction of molecular hydrogen/deuterium with atomic oxygen, \(V_ad^O+D_2\to OD+D(X)\) which is given in Fig. 8. Notice that the height of the adiabatic potential barrier is about 0.0015 Hartree, more then order of magnitude smaller in comparison to the height of the static potential barrier of 0.02 Hartree as shown in Fig. 6. It shows how the coupling between the vibrations perpendicular to the reaction coordinate to the kinetic energy component along the reaction coordinate enables the reactants to bypassing the static potential barrier to get the reaction products OD + D.

This adiabatic potential is exactly equal to the potential given in Eq. 6.3 and in Fig. 1c in ref. 37 for the O + D2 → OD + D reaction. Potential in Hartree and coordinate in atomic unites.
By using uniform complex scaling, where the X coordinate was rotated into the complex plane by an angle of θ = 0.75 rad15, the resonances for the adiabatic Hamiltonian for OD2 were calculated and counted by the number of their nodes. The results for the transition state (TS) and dynamical barrier (DB) resonances are given in Fig. 9. By comparing the static and adiabatic potentials presented in Figs. 6 and 8, it is clear that the complex poles in Fig. 9 are all associated with the dynamical barrier potential and not with the static potential barrier. Notice that the transition state is associated with the longest lifetime resonance, which is localized at the top of the adiabatic potential barrier (~0.0015 hartree). The other resonances (denoted as dynamical potential-barrier resonances) are associated with the excited vibrations that are perpendicular to the reaction coordinate. The cavity is tuned to couple the two states marked in blue in Fig. 9.
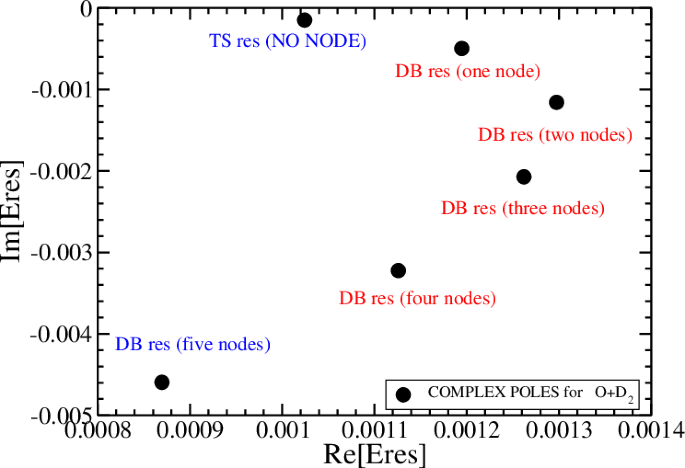
Energies in Hartree. The two resonances that will be coupled by the cavity are marked in blue.
The ability to enhance the rate of the reaction O + D2 → OD + D is due to the fact that there is a DB resonance (mark by a blue color) which its position is lower than the position of the TS resonance (mark also by a blue color). Therefore through emission of one photon the TS resonance will have the same energy as of the five nodes excited DB resonance. As we will show below in the next section the rate of this reaction is enhanced when the distance between the two mirrors is tuned to have a quantized field mode state with the energy ℏωcav which is equal to the energy gap between the TS and DB resonances. That is, ℏωcav = Re[Eres(TS) − Eres(DB)] = 0.0001547Hartree. The condition that Re[Eres(TS) − Eres(DB)] > 0 implies that due to the emission of one photon the two resonances becomes almost degenerate with the energy of the DB resonance (this resembles the situation in standard stimulated emission). In the next section we will describe how the two molecular states are mixed with the quantized radiation field modes to form upper and lower polaritons. This condition should be satisfied in any other reaction that its rate can be enhanced by a dark cavity.
link




.jpg "IFW Dresden selects Agnitron Agilis 100 MOCVD platform for precursor chemistry and ultra-wide-bandgap materials development")