
Bridging electron and nuclear motions in chemical reactions through electrostatic forces from reactive orbitals

Electrostatic forces exerted by electrons on nuclei
To understand the forces acting on nuclei during chemical reactions, we begin by examining the electrostatic forces exerted by all electrons on the nuclei within a molecule. The Hamiltonian, \(\hat \), governing the system of electrons and nuclei24, is given by
$$\hat =\sum\limits_^{ _{{{ }}}}\left(-\frac _^ -\sum\limits_{A}^{{n}_{{{{{\rm{nuc}}}}}}}\frac{{Z}_{A}}{{r}_{iA}}\right)+\sum\limits_{i < j}^{{n}_{{{{{\rm{elec}}}}}}}\frac{1}{{r}_{ij}}+\sum\limits_{A < B}^{{n}_{{{{{\rm{nuc}}}}}}}\frac{{Z}_{A}{Z}_{B}}{{R}_{AB}},$$
(1)
where \({\nabla }_{i}^{2}\) denotes the Laplacian with respect to electron i, riA is the distance from electron i to nucleus A, rij is the inter-electronic distance between electrons i and j, RAB represents the internuclear distance between nuclei A and B, ZA is the charge of nucleus A, and nelec and nnuc represent the numbers of electrons and nuclei, respectively. Atomic units are used (ℏ = e2 = m = 1, energies are in hartree, and distances are in bohr). Building upon electrostatic theory15, the Hellmann-Feynman force exerted by the electrons and nuclei on nucleus A is expressed as
$${{{{{\bf{F}}}}}}_{A} = -\frac{\partial }{\partial {{{{{\bf{R}}}}}}_{A}}\left\langle \Psi \left\vert \hat{H}\right\vert \Psi \right\rangle =-\left\langle \Psi \left\vert \frac{\partial \hat{H}}{\partial {{{{{\bf{R}}}}}}_{A}}\right\vert \Psi \right\rangle \\ \simeq {Z}_{A}\int\,d{{{{\bf{r}}}}}\,\rho ({{{{\bf{r}}}}})\frac{{{{{\bf{r}}}}}-{{{{{\bf{R}}}}}}_{A}}{| {{{{\bf{r}}}}}-{{{{{\bf{R}}}}}}_{A}{| }^{3}}-{Z}_{A}\sum\limits_{B(\ne A)}^{{n}_{{{{{\rm{nuc}}}}}}}{Z}_{B}\frac{{{{{{\bf{R}}}}}}_{AB}}{{{R}_{AB}}^{3}},$$
(2)
where ρ(r) represents the electron density at position r, and RA and RAB are position vector of nucleus A and vector from nucleus A to nucleus B, respectively. According to the Hellmann-Feynman theorem, these forces represent classical electrostatic forces when the electron distribution is determined variationally16. The approximation in the right-hand side of Eq. (2) neglects physical effects that depend on nuclear coordinates, such as dispersion forces, which scale with the inverse sixth power of the interatomic distance. It is known that while dispersion forces can affect the structures of weakly bound molecules, they have little influence on molecular orbitals. Therefore, their contribution can generally be neglected, except in cases where the Hellmann-Feynman force is extremely weak.
For a wavefunction describing all electrons, the electron density is given by
$$\rho ({{{{{\bf{r}}}}}}_{1})=N\int\,d{s}_{1}d{{{{{\bf{x}}}}}}_{2}\cdots d{{{{{\bf{x}}}}}}_{N}{\Psi }^{* }({{{{{\bf{x}}}}}}_{1},{{{{{\bf{x}}}}}}_{2},\cdots \,,{{{{{\bf{x}}}}}}_{N})\times \Psi ({{{{{\bf{x}}}}}}_{1},{{{{{\bf{x}}}}}}_{2},\cdots \,,{{{{{\bf{x}}}}}}_{N}),$$
(3)
which is integrated over all spatial and spin coordinates, xn = (rn, sn) for n = 1 to N, except r1. Within independent electron approximations25, such as the Kohn-Sham method26, the electron density simplifies to
$$\rho ({{{{\bf{r}}}}})=\sum\limits_{i}^{{n}_{{{{{\rm{elec}}}}}}}{\rho }_{i}({{{{\bf{r}}}}})=\sum\limits_{i}^{{n}_{{{{{\rm{elec}}}}}}}{\phi }_{i}^{* }({{{{\bf{r}}}}}){\phi }_{i}({{{{\bf{r}}}}}),$$
(4)
where ϕi is the i-th spin orbital wavefunction. The total electrostatic force on nucleus A is then expressed as the sum of contributions from electrons (\({{{{{\bf{F}}}}}}_{A}^{{{{{\rm{elec}}}}}}\)) and other nuclei (\({{{{{\bf{F}}}}}}_{A}^{{{{{\rm{nuc}}}}}}\)),
$${{{{{\bf{F}}}}}}_{A}={{{{{\bf{F}}}}}}_{A}^{{{{{\rm{elec}}}}}}+{{{{{\bf{F}}}}}}_{A}^{{{{{\rm{nuc}}}}}}\simeq {Z}_{A}\sum\limits_{i}^{{n}_{{{{{\rm{elec}}}}}}}{{{{{\bf{f}}}}}}_{iA}-{Z}_{A}\sum\limits_{B(\ne A)}^{{n}_{{{{{\rm{nuc}}}}}}}{Z}_{B}\frac{{{{{{\bf{R}}}}}}_{AB}}{{{R}_{AB}}^{3}},$$
(5)
where the force contribution from the i-th orbital on nucleus A is
$${{{{{\bf{f}}}}}}_{iA}=\int\,d{{{{\bf{r}}}}}{\phi }_{i}^{* }({{{{\bf{r}}}}})\frac{{{{{\bf{r}}}}}-{{{{{\bf{R}}}}}}_{A}}{| {{{{\bf{r}}}}}-{{{{{\bf{R}}}}}}_{A}{| }^{3}}{\phi }_{i}({{{{\bf{r}}}}}),$$
(6)
Using this framework, the influence of reactive orbitals on nuclear forces can be isolated. The force contribution from an occupied reactive orbital (ORO) is given by
$${{{{{\bf{f}}}}}}_{A}^{{{{{\rm{ORO}}}}}}=\int\,d{{{{\bf{r}}}}}{\phi }^{{{{{\rm{ORO* }}}}}}({{{{\bf{r}}}}})\frac{{{{{\bf{r}}}}}-{{{{{\bf{R}}}}}}_{A}}{| {{{{\bf{r}}}}}-{{{{{\bf{R}}}}}}_{A}{| }^{3}}{\phi }^{{{{{\rm{ORO}}}}}}({{{{\bf{r}}}}}),$$
(7)
where ϕORO represents the wavefunction of the ORO. This formulation allows for the direct assessment of how variations in reactive orbitals influence nuclear forces during chemical reactions.
In this study, we investigate the primary electrostatic forces driving nuclear motions in chemical reactions, focusing specifically on the forces generated by variations in the ORO. The progression of electron transfer throughout a reaction is characterized by changes in OROs13. The electrostatic force vector resulting from ORO variations can be expressed as:
$${{{{{\bf{F}}}}}}_{A}^{{{{{\rm{ROEF}}}}}}={Z}_{A}{{{{{\bf{f}}}}}}_{A}^{{{{{\rm{ORO}}}}}}.$$
(8)
where we refer to \({{{{{\bf{F}}}}}}_{A}^{{{{{\rm{ROEF}}}}}}\) as the reactive orbital-based electrostatic force (ROEF) vector. This formulation isolates the contribution of ORO variations along the reaction pathway, which may involve interactions with unoccupied orbitals, while neglecting the contributions from other orbitals.
Relationship between electrostatic forces and orbital energies
Next, we examine the relationship between ROEF and the Kohn-Sham orbital energies24. The Kohn-Sham orbital energy, ϵi, is defined as
$${\epsilon }_{i}={h}_{i}+\sum\limits_{j}^{{n}_{{{{{\rm{elec}}}}}}}{J}_{ij}+\int\,d{{{{\bf{r}}}}}{\rho }_{i}({{{{\bf{r}}}}}){v}_{{{{{\rm{xc}}}}}},$$
(9)
where vxc is an exchange-correlation potential functional, and hi represents the one-electron Hamiltonian, given by
$${h}_{i}=\int\,d{{{{\bf{r}}}}}{\phi }_{i}^{* }({{{{\bf{r}}}}})\left\{-\frac{1}{2}{\nabla }^{2}-\sum\limits_{A}^{{n}_{{{{{\rm{nuc}}}}}}}\frac{{Z}_{A}}{{r}_{iA}}\right\}{\phi }_{i}({{{{\bf{r}}}}}).$$
(10)
The derivative of the orbital energy with respect to the coordinates of nucleus A is then expressed as
$$\frac{\partial {\epsilon }_{i}}{\partial {{{{{\bf{R}}}}}}_{A}}=\frac{\partial }{\partial {{{{{\bf{R}}}}}}_{A}}\left\{{h}_{i}+\sum\limits_{j}^{{n}_{{{{{\rm{elec}}}}}}}{J}_{ij}+\int\,d{{{{\bf{r}}}}}{\rho }_{i}({{{{\bf{r}}}}}){v}_{{{{{\rm{xc}}}}}}\right\}=\frac{\partial {h}_{i}}{\partial {{{{{\bf{R}}}}}}_{A}}.$$
(11)
where contributions from the vxc term are negligible due to its limited explicit dependence on nuclear positions.
Reactive-orbital-based electrostatic force (ROEF)
The total electrostatic force vector on nucleus A, derived from the Kohn-Sham electronic energy, EKS, can be written as
$${{{{{\bf{F}}}}}}_{A}^{{{{{\rm{elec}}}}}} =-\frac{\partial {E}_{{{{{\rm{KS}}}}}}}{\partial {{{{{\bf{R}}}}}}_{A}}=-\frac{\partial }{\partial {{{{{\bf{R}}}}}}_{A}}\left\{\sum\limits_{i}^{{n}_{{{{{\rm{elec}}}}}}}{h}_{i}+\sum\limits_{i}^{{n}_{{{{{\rm{elec}}}}}}}\sum\limits_{j(\ne i)}^{{n}_{{{{{\rm{elec}}}}}}}{J}_{ij}+{E}_{{{{{\rm{xc}}}}}}\right\}\\ =-\sum\limits_{i}^{{n}_{{{{{\rm{elec}}}}}}}\frac{\partial {h}_{i}}{\partial {{{{{\bf{R}}}}}}_{A}}=-\sum\limits_{i}^{{n}_{{{{{\rm{elec}}}}}}}\frac{\partial {\epsilon }_{i}}{\partial {{{{{\bf{R}}}}}}_{A}},$$
(12)
indicating that the force contribution from each orbital is proportional to its energy gradient with respect to nuclear coordinates. Substituting this relationship, the ROEF vector can be expressed as
$${{{{{\bf{F}}}}}}_{A}^{{{{{\rm{ROEF}}}}}}=-\frac{\partial {\epsilon }^{{{{{\rm{ORO}}}}}}}{\partial {{{{{\bf{R}}}}}}_{A}},$$
(13)
where ϵORO is the orbital energy of the ORO. This equation directly relates the orbital energy gradients to the forces exerted on the nuclei, providing a quantitative link between electronic structure and nuclear motion. By focusing on the ORO, this approach establishes a direct connection between orbital energy variations and nuclear forces, enabling a more detailed understanding of the forces driving chemical reactions at the atomic level.
Accuracy and interpretation of ROEF
From the relationship established in Eq. (13), we infer that orbital energy variations influence electrostatic forces during chemical reactions. Specifically, for OROs that exhibit decreasing orbital energies as the reaction progresses, the resulting electrostatic forces are expected to align with the reaction direction, as described in Eq. (8). These reaction-aligned electrostatic forces create trenches on the PES, delineating the intrinsic reaction coordinate (IRC) and defining the reaction pathway. OROs act as the driving force for a reaction when their electrostatic forces are sufficiently strong to establish the IRC. Since orbital energy remains constant during idealized electron transfer27, orbital energy variations are minimal in reaction stages dominated by electron transfer. This behavior is reflected in the early stages of many reactions, as shown in Figs. S1 and S2 of the Supplementary Information. According to Eq. (13), when the orbital energy gradient approaches zero, the corresponding electrostatic force vector vanishes. However, even small gradients in orbital energy, arising from structural transformations along the IRC, determine the magnitude of the electrostatic force. Accurate calculation of these forces, therefore, hinges on precise determination of orbital energy variations induced by structural changes. This underscores the necessity of a robust theoretical framework capable of reliably predicting orbital energy gradients across different molecular structures. LC-DFT provides a critical tool in this regard, as it quantitatively reproduces orbital energies with high fidelity, making it indispensable for such analyses.
The Pulay force, an artificial force arising from the use of basis sets, is a factor potentially related to ROEFs28,29,30. Previous studies report that the magnitude of Pulay forces for all electrons is less than 10 kJ mol−1 bohr−1, with even smaller contributions for individual molecular orbitals. By contrast, ROEF derived from orbital energy gradients are on the order of several hundred kJ mol−1 bohr−1. Given the negligible magnitude of Pulay forces compared to ROEFs, they are excluded from this analysis for simplicity.
An important insight from the interplay between ROEFs and orbital energy variations relates to the virial theorem31, which states that the kinetic energy is half the potential energy but with the opposite sign for nuclear-electron interactions. Under independent electron approximations, this relationship holds for individual electrons32. When orbital energy variations during orbital mixing are small, nuclear-electron potential changes are similarly minimized, suppressing fluctuations in ROEFs, as shown in Eqs. (10) and (12). While electron transfer induces polarization in the electron distribution, ROEF remains stable. However, structural deformations near the transition state can generate significant ROEFs, influencing the reaction pathway.
This study highlights the critical role of electron transfer as the driving force for structural transformations during reactions. By calculating the electrostatic forces associated with ORO energy gradients along the IRC, we trace the forces exerted on atomic nuclei, revealing how changes in electron distribution induced by electron transfer shape the reaction pathway.
Three-body model for evaluating ROEFs
Using the framework of ROEFs, we analyze a diverse set of atom-transfer reactions, comprising 16 hydrogen transfer reactions, one heavy atom transfer, two nucleophilic substitutions, and five unimolecular reactions, for the forward and backward processes, based on their calculated IRCs. For the ROEF analysis, atom-transfer reactions were modeled using a three-body approach.
Figure 1 illustrates this framework, applied to the NH + CH4 → NH2 + CH3 reaction (a) and the ROEF vector calculation method (b). These ROEF values provide insight into the forces driving atomic movement caused by electron redistribution during ORO variations. This three-body model provides a simple and intuitive way to visualize the correlation between the reaction direction and electrostatic forces. However, it should be noted that this model is not applicable to more complex reactions such as catalytic or enzymatic processes, which are beyond the scope of the present study. To visualize such correlations in complex reactions, a new model will be required—one that projects the electrostatic force vectors onto the nuclear displacement vectors along the reaction coordinate. ROEF variations along the IRCs for all calculated reactions, along with corresponding molecular structures and atomic coordinates, are presented in Figs. S1 and S2 of the Supplementary Information.
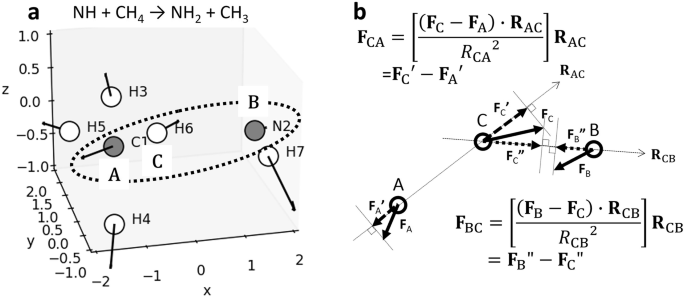
a Example calculation model for the NH + CH4 → NH2 + CH3 reaction at the transition state, where atoms C1, N2, and H6 are labeled as A, B, and C, and the movement of atom C from A to B is considered. b Method for calculating ROEFs in atom-transfer reactions: FA, FB, and FC are the ROEF vectors calculated for atoms A, B, and C, respectively, \({{{{{\bf{F}}}}}}_{{{{{\rm{A}}}}}}^{{\prime} }\) and \({{{{{\bf{F}}}}}}_{{{{{\rm{C}}}}}}^{{\prime} }\) are the projection vectors of FA and FC in the direction of the A-C bond, FC” and FB” are the projection vectors of FC and FB in the direction of the C-B bond. These projections enable the calculation of ROEF vectors, FBC and FCA, crucial to the reaction. For unimolecular reactions, only one ROEF vector set corresponds to the reaction direction, and the remaining vectors are set to zero.
link




.jpg "IFW Dresden selects Agnitron Agilis 100 MOCVD platform for precursor chemistry and ultra-wide-bandgap materials development")